Contents
- Introduction
- Clinical Characteristics of Cerebral Edema Complicating DKA
- The Osmotic Abnormalities in DKA
- Overhydration and Hyponatremia During DKA
- Summary and Conclusions
- Suggested Readings
- Clinical Characteristics of Cerebral Edema Complicating DKA
Introduction
In 1922 Banting and Best introduced insulin into clinical practice. A decade later the first reported case of cerebral edema complicating diabetic ketoacidosis (DKA) was reported by Dillon, Riggs and Dyer writing in the pathology literature. While the syndrome of cerebral edema complicating DKA was either not seen, ignored, or was unrecognized by the medical community until 3 decades later when the complication was again reported by Young and Bradley at the Joslin Clinic, there has since been a flurry of case reports in the 1960's and 1970's and basic and clinical research from the 1970's to the 1990's leading to our present day acceptance of this as a known complication of DKA, or of the management of DKA.
In fact, we now recognize that the cerebral complications of DKA (including much less frequent cerebral arterial infarctions, venous sinus thrombosis, and central nervous system infections) are the most common cause of diabetic-related death of young diabetic patients (1), accounting for 31% of deaths associated with DKA and 20% of all diabetic deaths, having surpassed aspiration, electrolyte imbalance, myocardial infarction, etc.
Furthermore, diabetes mellitus remains an important cause of hospitalization of young children. The prevalence rate of diabetes continues to grow in all Western developed nations, nearly doubling every decade, resulting in 22,000 hospital admissions in children under 15 years of age for diabetes in the United States in 1994, the majority of which were due to ketoacidosis. With approximately 4 hospital admissions of children for DKA per 100,000 population per year (2), every PICU located in a major metropolitan center will continue to see children with DKA at significant rates in the future, and every pediatric intensivist will see at least one case of cerebral edema complicating DKA during his/her career.
The Clinical Characteristics of Cerebral Edema Complicating DKA
Cerebral edema complicating DKA is a syndrome unique to pediatrics. While described in a few adults, this complication occurs predominantly in children, most of whom are experiencing their onset of diabetes mellitus with the presentation of DKA (3) (Figure 1).
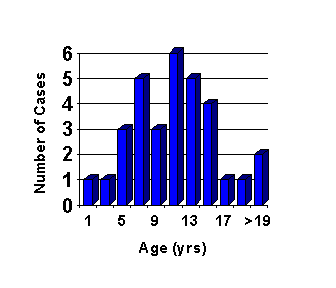
Figure 1. The age of patients in approximately 30 cases of cerebral edema complicating DKA from the author's case files and that were reported in the literature between 1960 and 1980.
Cerebral edema complicating DKA is generally an unpredictable phenomenon that occurs in children who seem to be metabolically returning to normal, generally 3-12 hours after the initiation of therapy (4-6) (Figure 2).
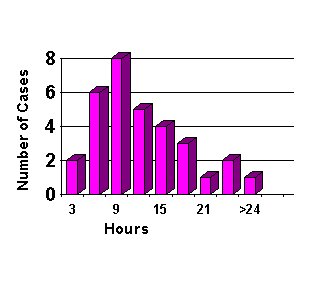
Figure 2. The time of onset of neurologic changes in approximately 30 cases of cerebral edema complicating DKA from the author's case files and that were reported in the literature between 1960 and 1980.
This description may seem true for the clinician faced with a case of cerebral edema. But as cases are subjected to post hoc scrutiny, and hindsight is liberally applied, it often seems to be the case that neurologic signs, while subtle, sometimes preceded the onset of clinically obvious cerebral edema and neurologic collapse. These symptoms may be increased lethargy and diminished arousability, incontinence, or complaints of headache. On the other hand, many of these complaints or observations may not seem remarkable at the time they occur. Lethargy and complaints of headache in the early hours of morning in a child with an acute viral illness, and who has been vomiting for several days, are truly not unusual, and may only be judged to be significant with the benefit of hindsight.
While it is true that only a small percent (0.5%-3%) of children with DKA will experience catastrophic cerebral edema, several investigations demonstrate that subclinical cerebral edema is common if not universal during treatment of DKA both in adults (7,8) and children (6). These observations of cerebral edema during treatment were expanded by Hoffman, et al. (9,10) who demonstrated that the radiologic and transcranial Doppler evidence of cerebral edema is present before the initiation of therapy for DKA.
We therefore recognize that cerebral edema is a common subclinical complication of DKA in children, and that in a very few unfortunate children the cerebral edema becomes clinically manifest, usually with devastating consequences. The cause of the underlying cerebral edema in DKA, and the cause of neurologic deterioration in the minority of children whose cerebral pathology progresses, have been the source of considerable debate over the years. The theories commonly invoked are several:
1. Osmotic dysequilibrium between brain and plasma.
2. Overhydration and hyponatremia.
3. Intracerebral acidosis induced by alkali therapy or the correction of acidosis; accumulation of intracellular Na due to the action of the Na-H membrane exchanger, the antiport (11-13).
4. Alterations in cerebral blood flow (9,14).
This review will discuss the putative role of theories 1 and 2 in the development of cerebral edema during DKA. References pertaining to theories 3 and 4 are provided for those interested in further reading.
The Osmotic Abnormalities in DKA
Several profound metabolic abnormalities exist in the child with DKA; among the most important are disorders in serum glucose and sodium concentrations, which are the principal determinants of secondary changes in the osmolality, osmolarity, and tonicity of the plasma. Because these three terms mean different things but are frequently used interchangeably, it is useful to define them.
By convention, many biological measurements of concentration are expressed as percentages, or as weight in grams per 100 ml of solution. For example, 0.9% saline solution contains 0.9 gm of NaCl per 100 ml of water, 5% dextrose contains 5 gm of glucose per 100 ml of water, and a blood glucose measurement of 80 mg-% means that there are 80 mg of glucose per 100 ml of blood. The accuracy of this measurement relies on laboratory accounting for the solution temperature at the time of measurement.
A measurement that is more biologically appropriate is the molal measurement of concentration, that is, the concentration in moles, millimoles, etc. per 1000 g of solvent, expressed as a lower case "m" (m, mm, µm, etc.). This measurement is independent of temperature. Alternatively, concentration of solute can be expressed in molar measurement, the number of moles, millimoles, etc. per liter of solution, expressed as an upper case "M" (M, mM, etc.). Because solution volume changes with temperature, molar concentration is temperature dependent.
Because biologic membranes are readily permeable to water molecules there is continual movement or exchange of water between various fluid compartments. The forces governing the movement of water are hydrostatic and osmotic pressures. Most important to this discussion is the latter. Quantitatively, osmotic pressures greatly exceed hydrostatic pressures in the body; the magnitude can be appreciated when one realizes that an osmotic pressure difference of only 6 mOsm/L can move as much water as the entire hydrostatic pressure generated by the heart.
Osmotic pressure is a property of solutions that depends on the number of osmotically active molecules in solution, not upon the ionic charge of those molecules, the size of the molecules, or the physical-chemical properties of the molecules. Thus 1 gram-molecular weight (1 mole) of a compound (such as glucose) is termed 1 osmole (osmol, Osm). Most biological concentrations are expressed in milliosmolar (mOsm) or microOsmolar (µOsm) concentrations. Ionized molecules (e.g. NaCl) dissociate in solution, and each dissociated ion exerts its own osmotic force. For example, NaCl is principally dissociated in the body, therefore 1 mM of NaCl exerts approximately 2 mOsm of osmotic pressure; 1mM of CaCl2 exerts 3 mOsm of osmotic pressure, etc. Osmolality is the expression of this quantity of osmotically active particles per 1000 gm of water or solvent, while the more commonly used osmolarity expresses the number of osmotically active particles per liter of solvent.
There is a small difference between osmolality and osmolarity principally due to dissolved protein and fat which comprise 6% to 8% of the solutes in plasma, but the difference is negligible when dealing with small concentrations of solute. In general, the serum osmolarity can be estimated to within 10% accuracy by the equation:
Sosm = 2 x [Na+] + [glucose]/18 + [BUN]/2.8
However, this equation will underestimate osmolarity in the presence of unusual or unmeasured solutes, such as ketoacids, ketone, and amino acids, all of which may be significantly elevated during DKA.
Tonicity is a term that is used to describe the relative osmolality of solutions; thus a solution is isotonic when it is isosmotic with body fluids. Normal saline and D5W are isotonic with respect to plasma, in that red blood cells will neither expand nor shrink if suspended in those solutions. Hypertonic fluids (5% NaCl, mannitol, etc.) have a greater solute concentration than body fluids, and will cause cellular dehydration and shrinkage due to egress of water from cells. Although the terms tonicity and osmolarity are often interchanged in clinical usage, an alteration of one does not necessarily imply an alteration of another. Tonicity, in this way, can be thought of as the effective osmolality (Eosm). High concentrations of low molecular weight permeant molecules (urea, ethanol) distribute evenly throughout total body water, and therefore produce no effect on tonicity. Uremia is a common example of a hyperosmolar state but not a hypertonic state. Glucose, however, is not a permeant molecule, therefore hyperglycemia indeed induces a hypertonic state. The effective osmolality or tonicity, can be best estimated using only glucose and sodium in the equation above (plus other exogenous impermeant solutes if any, such as mannitol, glycerol, etc.):
Eosm = 2 x [Na+] + [glucose]/18
DKA clearly represents a hypertonic state, as glucose is significantly elevated in concentration in the plasma. It was recognized several decades ago, in studies of hypernatremic dehydration, that over-rapid correction of hypertonic states could lead to cerebral swelling and death. This phenomenon was attributed to the development or generation of what was then termed "idiogenic osmoles," osmotically active substances formed within the cells of the brain that served to counter the extra osmolality of plasma and protect the cells of the brain from shrinkage during episodes of hypertonicity. Experimental animals made hypertonic with exogenously administered compounds (sodium, glucose, mannitol) had concomitant increases in brain osmolality (15). The term idiogenic osmoles was coined because of the inability of investigators to chemically identify these new osmotically active material in the brains of hypertonic experimental animals (Figure 3) (15,16).
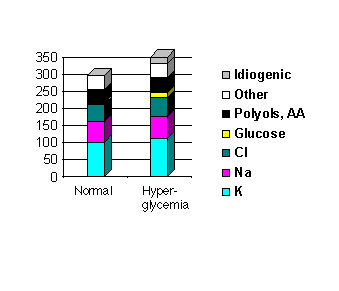
Figure 3. Animals made hypertonic have an increase in brain osmolality; the left column represents the osmotically active constituents of a normal rabbit brain; the right column represents the same in a rabbit made hyperglycemic (glucose 1000 mg/dl). The brain osmolality increased both because of a general increase in electrolyte and glucose concentrations, but also because of the generation of about 20 mOsm of unknown osmoles, termed "idiogenic osmoles." (Adapted from Arieff & Kleeman, 1974)
It was not a very large leap to then postulate that these idiogenic osmoles, putatively the result of intra-cerebral glucose metabolism and therefore large molecules, would dissipate slowly leaving the brain hypertonic relative to plasma during restoration of euglycemia during treatment of DKA, and rendering it susceptible to swelling as water moved into the brain along its osmotic gradient (Figure 4). Indeed, idiogenic osmoles are present in experimental animals during experimental hyperglycemia and during induced DKA. The identity of at least some of these osmoles has been established as myoinositol(~1 mOsm/kg), taurine(~3.5 mOsm/kg), and urea (the last, of course, an osmole but not one participating in brain tonicity for reasons discussed above) (15,17).
Figure 4. The theory of idiogenic osmoles producing brain edema:
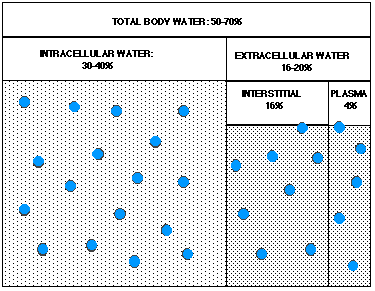
Fig. 4a. Normal body water compartments. Dots denote sodium ions, and large dots denote glucose molecules.
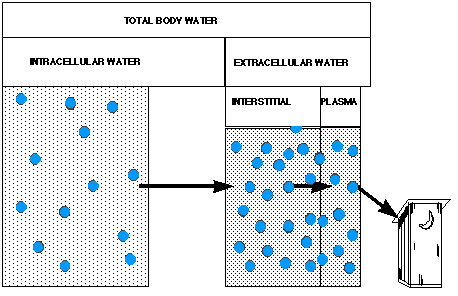
Fig. 4b. During hyperglycemia, extracellular glucose concentration rises, leading to a preferential loss of water from the intracellular compartment. Water molecules travel in the direction of the arrows, and ultimately are lost in an osmotic diuresis.
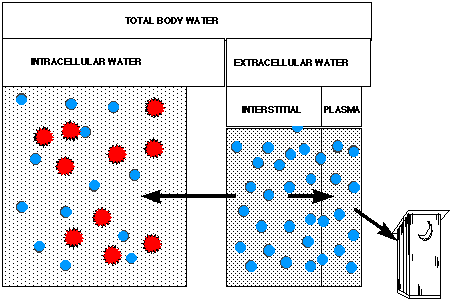
Fig. 4c. Later during hyperglycemia, the brain generates "idiogenic osmoles" (denoted by stars) which serve to draw water back into the cerebrum, ameliorating intracellular dehydration.
But what is the evidence that a reduction of hyperglycemia and correction of hypertonicity causes cerebral edema?
The seminal work in this area was completed by Arieff and colleagues, who were unable to establish such a link. In their rabbit model, brain osmolality increased with increasing magnitude and duration of hyperglycemia, by as much as 40 mOsm/kg during 4 hours of hyperglycemia to levels of about 1000 mg/dl. Half of the increase in brain osmolality was accounted for by increases in intracellular ion and glucose concentration, and half of which by the generation of "idiogenic osmoles." However, they noted "when plasma glucose is . . . lowered below 14 mM, cerebral edema occurs which is characterized by a significant augmentation in the cerebral cortex content of Na+, K+, Cl- and water. Idiogenic osmoles are not only retained in the brain under such circumstances, but the quantity which are present during hyperglycemia increases still further as the plasma glucose is lowered towards normal with insulin. By contrast, when the plasma glucose and Osm are lowered by means of peritoneal dialysis and hypotonic saline infusion, an osmotic gradient between brain and plasma does not develop." In other words, experimental brain swelling occurs with the correction of hyperglycemia with insulin, but does not occur with rapid correction of hyperglycemia with dialysis and hypotonic fluids, and without the use of insulin. Arieff, et al. were led to speculate on the role of insulin on membrane ion pumps, and to caution not to correct hyperglycemia below 250 mg/dl.
Strikingly similar results were found by Tornheim, who studied the brains of diabetic rats treated with insulin or fluid alone to correct DKA (18). She wrote, "Treatment with saline alone did not result in central overhydration. The findings of this study suggest that aggressive therapy with fluids and insulin, but not fluid alone, results in global overhydration of the brains of diabetic animals. Prolonged fluid and insulin treatment with a return of blood glucose to normal values causes further and preferential accumulation of edema fluid in the cerebral cortex."
How may we integrate this knowledge into clinical practice, and how does it reconcile with clinical experience? On balance, one may safely conclude that the osmolality of the brain increases during untreated episodes of hyperglycemia, due to an influx of cations, glucose, and the generation of larger complex carbohydrates and amines. However, these substances dissipate rapidly and brain edema does not occur if hyperglycemia is corrected with even massive quantities of isotonic (18) or hypotonic (15) fluids; if however hyperglycemia is treated with a combination of fluids and insulin, the brain accumulates further cations and other osmotically active substances, and brain edema occurs. Because all children with DKA are treated with insulin, the data are consistent with the observation that cerebral edema occurs only after the initiation of therapy (with the exception of a few rare case reports). These data are also consistent with the observation that cerebral edema is a very rare complication of nonketotic hyperglycemic coma (NKHC), even though NKHC is associated with hyperglycemia many times more severe than DKA. NKHC is generally treated with fluid resuscitation alone, but not insulin; in fact, insulin therapy has been associated with cerebral edema in this setting (19). These data cannot, however, be easily reconciled with the several observations of ICP, radiographic, and encephalographic evidence of brain edema before the onset of therapy with fluids or insulin.
Overhydration and Hyponatremia During DKA
As clinicians have sought to understand cerebral edema complicating DKA and identify strategies for preventing it, considerable attention has been turned towards the possible role of fluid therapy while all but ignoring a potential role of insulin in the etiology of cerebral edema.
The first call to arms against aggressive rehydration of children with DKA came from Duck, et al. in 1976 (19), who described 4 cases of cerebral edema and combined their data with the data of another 5 previously reported in the literature. These authors observed that all of the children who developed cerebral edema had received fluid therapy at a rate in excess of 4 L/m2/day during the first 3-15 hours of therapy. Compared to an unmatched group of 21 children from these authors' practice who did not develop cerebral edema, these 4 children had lower average serum sodium concentrations measured at the same hour that cerebral edema occurred (124 vs 131 mEq/L). Unfortunately, the authors do not describe the fluid therapy of this cohort group of 21 children.
Remember, however, that protocols in practice throughout the 1970's called for fluid resuscitation of diabetic patients to be proportional to their degree of dehydration. Most protocols called for initial fluid boluses of 10-30 ml/kg followed by replacement of half the fluid deficit over the first 8 hours of therapy. If we calculate what this would mean for a 30 kg 0.7 m2 child with "typical" DKA, that is, who is 10%-12% dehydrated, we would prescribe a fluid bolus of 500 ml of isotonic fluid followed by an infusion of hypotonic fluid at 145 ml/hr for a 8 hour fluid total of 1.65 L, half his deficit. This amounts to 2.4 L/m2/8 hr, or 7 L/m2/24 hr, well above the Duck threshold for risk of cerebral edema. In other words, it is hardly surprising that all of Duck's patients with cerebral edema received fluid in excess of 4 L/m2/8 hr because virtually all protocols recommended this for all but the minimally dehydrated child. Duck's analysis, therefore, fails to take into account the severity of DKA and dehydration as a factor in the causality of cerebral edema, and presents us only with a "true-true-unrelated" paradigm.
A decade later the same senior author expanded his observations of the rate of rehydration and the incidence of hyponatremia in the etiology of cerebral edema (20) reporting 9 new cases and comparing them to 39 previously published reports. Without dwelling on the inherent weaknesses of retrospective series, especially ones in which the majority of data is gleaned from secondary sources (published reports) rather than medical records, their findings viewed objectively may actually weaken their argument of fluid management as a cause of cerebral edema, and hyponatremia as a contributing cause or marker. In this series, 10% of cases of cerebral edema occurred following fluid resuscitation at less than Duck's threshold of 4 L/m2/24 hr, again, not surprising given published protocols for the management of DKA. Two-thirds of his reported subjects did not have measured serum sodiums below 130 mEq/L at the time of neurologic collapse, although many had declining sodium values. Once again, Duck did not distinguish between rapid rehydration as an etiology of cerebral edema, versus an independent cause of cerebral edema in the more severely dehydrated children, who received more rapid rehydration by protocol to manage their greater degree of dehydration.
The other author who has written in favor of conservative fluid management is Harris, et al. (21,22). In his earlier paper, Harris retrospectively studied over 200 episodes of DKA in children and adults, and described his prospective experience with a slow-rehydration regimen in 58 subsequent patients. In the former retrospective group, he identified 20 neurologic complications noted as minor (headache, n=13) and major (death or neurologic collapse, n=7), and noted that serum sodium failed to increase in 54% of uncomplicated cases of DKA, but in 95% of so-defined complicated cases (p<0.01). In discussing his prospective cohort group who were rehydrated over 48 hrs, he noted that sodium rose in 95% of patients, and no "major" complications occurred. While the behavior of serum sodium was impressive, the absence of complication (even if defined as a headache, and assumed to be 0 in incidence in the slow rehydration group) did not reach statistical significance in comparing the rapid to slow rehydration groups (p=0.4 by chi-square calculated from their data).
In the latter Harris paper, the authors describe a 5 year experience of 48 hour hydration in 231 episodes of DKA. They stratified their subjects into mildly and severely dehydrated groups, and observed that there was a statistically significant difference between the groups in admission glucose, BUN, and sodium. They had no major neurologic complications with their treatment protocol, concluding that "gradual rehydration can protect against life-threatening increases in intracranial pressure and brain herniation."
The Harris conclusions that slow rehydration is safe, vis a vis resolution of the abnormalities of DKA, is probably correct, however can one conclude that slow rehydration protects against cerebral edema? To answer this question I would refer the reader to an important and simple biostatistics paper, "If Nothing Goes Wrong, Is Everything All Right?" (23) in which Hanley demonstrates that a zero numerator in a clinical study does not imply zero risk, even with large denominators, but that the 95% confidence interval for stating a 0/n rate is 3/n. In other words, in the Harris study one can state that with 48 hour rehydration the risk of cerebral edema is not larger than 0 + 3/231, or 1.2%, approximately the same magnitude of risk already documented in prior studies without control of rehydration rates. One can hardly argue with the call for prudent fluid therapy and careful monitoring for and management of hyponatremia, but it is premature to state that this will prevent cases of cerebral edema.
The role of hyponatremia is also uncertain. Hyponatremia may play a role in promoting the development of cerebral edema, or it may be a marker for children who are developing cerebral pathology and are manifesting a degree of SIADH on their way to a neurologic catastrophe. Hyponatremia is far from a constant occurrence during the development of DKA: The original case reports of cerebral edema during DKA from the 1960's described children who were hypernatremic, and the Duck and Harris papers describe hyponatremia with varying frequency. Certainly it's difficult to assign a definite etiologic role to a phenomenon which often is not seen during the evolution of cerebral edema.
Finally, several other authors have retrospectively sought an association between fluid therapy, hyponatremia, and cerebral edema, but have failed to demonstrate such a link (3,24). In fact, cerebral edema has been reported in children who received only oral rehydration and insulin (25). The etiologic role of fluid resuscitation is therefore unresolved.
Summary and Conclusions
Severe metabolic and osmolal disturbances occur during routine episodes of DKA, and the latter has been associated with the development of increased cerebral osmolality that has been assumed to be responsible for the development of cerebral edema. However, it is possible to substantially correct hyperglycemia by administering massive quantities of fluid without inducing cerebral edema, and in so doing cerebral idiogenic osmoles dissipate.
Efforts by clinicians to identify a consistent etiology in patients with DKA who develop cerebral edema have not been successful. Neither excessive fluid therapy nor hyponatremia are constant features of the disease. Of some interest is the rarity of cerebral edema during treatment of the much more severe hyperglycemic states during NKHC, while its more frequent occurrence during the management of DKA.
The one common denominator of children who develop cerebral edema complicating DKA is therapy with insulin, and insulin has been experimentally shown in more than one study to be associated with the accumulation of cations and complex carbohydrates in the brain, and the development of cerebral edema during correction of hyperglycemia in laboratory animals. Insulin may have a direct ion gate effect on glial cells and neurons, or alternatively may act indirectly by accelerating the correction of metabolic acidosis associated with DKA, and thus activating a pH sensitive sodium-proton membrane pump.
Finally, the observation of the presence of subclinical cerebral edema before the initiation of therapy of DKA, and its worsening and persistence during management, is interesting and difficult to integrate into present theories regarding the etiology of cerebral edema.
The calls for careful management of serum sodium and conservative fluid management with a deceleration of fluid resuscitation from over 24 hours to over 48 hours are prudent and cannot be criticized, but it will be several years before sufficient clinical data may be accumulated to determine if such conservative management will reduce the incidence of this frequently devastating and incompletely understood complication of childhood diabetes. Until we understand more about its prevention, then we must remain vigilant to its occurrence and treat cerebral edema aggressively with conventional neuro-intensive care measures to lower ICP and restore cerebral blood flow.
Suggested Readings
1. Scibilia J, Finegold D, Dorman J, Becker D, Drash A. Why do children with diabetes die? Acta Endocrinol Suppl. 1986; 279:326-33.
2. Graves EJ, Gillum BS. 1994 Summary: National Hospital Discharge Survey. Advance data from vital and health statistics. 1996; 278:1-12, National Center for Health Statistics.
3. Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care. 1990; 13:22-33.
4. Bratton SL, Krane EJ. Diabetic Ketoacidosis: Pathophysiology, Management and Complications. J Intensive Care Med. 1992; 7:199-211.
5. Krane EJ. Diabetic ketoacidosis. Biochemistry, physiology, treatment, and prevention. Pediatr Clin North Am. 1987; 34:935-60.
6. Krane EJ, Rockoff MA, Wallman JK, Wolfsdorf JI. Subclinical brain swelling in children during treatment of diabetic ketoacidosis. N Engl J Med. 1985; 312:1147-51.
7. Clements RS, Jr., Blumenthal SA, Morrison AD, Winegard AI. Increased cerebrospinal fluid pressure during therapy for diabetic acidosis. Trans Assoc Am Physicians. 1971; 84:102-12.
8. Fein IA, Rachow EC, Sprung CL, Grodman R. Relation of colloid osmotic pressure to arterial hypoxemia and cerebral edema during crystalloid volume loading of patients with diabetic ketoacidosis. Ann Intern Med. 1982; 96:570-5.
9. Hoffman WH, Pluta RM, Fisher AQ, Wagner MB, Yanovski JA. Transcranial Doppler ultrasound assessment of intracranial hemodynamics in children with diabetic ketoacidosis. J Clin Ultrasound. 1995; 23:517-23.
10. Hoffman WH, Steinhart CM, el Gammal T, Steele S, Cuadrado AR, Morse PK. Cranial CT in children and adolescents with diabetic ketoacidosis. Am J Neuroradiol. 1988; 9:733-9.
11. Van der Meulen JA, Klip A, Grinstein S. Possible mechanism for cerebral oedema in diabetic ketoacidosis. Lancet. 1987; 2:306-8.
12. Mellergard P, Ouyang YB, Siesjo BK. The regulation of intracellular pH is strongly dependent on extracellular pH in cultured rat astrocytes and neurons. Acta Neurochir Suppl (Wien). 1994; 60:34-7.
13. Wuttke WA, Walz W. Sodium- and bicarbonate-independent regulation of intracellular pH in culture mouse astrocytes. Neurosci Lett. 1990; 117:105-10.
14. Quyyumi AA, Iaffaldano R, Guerrero JL, Ryan CA, Powell WJ, Axelrod L. Prostacyclin and pathogenesis of hemodynamic abnormalities of diabetic ketoacidosis in rats. Diabetes. 1989; 38:1585-94.
15. Arieff AI, Kleeman CR. Studies on mechanisms of cerebral edema in diabetic comas. Effects of hyperglycemia and rapid lowering of plasma glucose in normal rabbits. J Clin Invest. 1973; 52:571-83.
16. Arieff AI, Kleeman CR. Cerebral edema in diabetic comas. II. Effects of hyperosmolality, hyperglycemia and insulin in diabetic rabbits. J Clin Endocrinol Metab. 1974; 38:1057-67.
17. Harris GD, Lohr JW, Fiordalisi I, Acara M. Brain osmoregulation during extreme and moderate dehydration in a rat model of severe DKA. Life Sci. 1993; 53:185-91.
18. Tornheim PA. Regional localization of cerebral edema following fluid and insulin therapy in streptozotocin-diabetic rats. Diabetes. 1981; 30:762-6.
19. Arieff AI. Cerebral edema complicating nonketotic hyperosmolar coma. Miner Electrolyte Metab. 1986; 12:383-9.
20. Duck SC, Wyatt DT. Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr. 1988; 113:10-4.
21. Harris GD, Fiordalisi I. Physiologic management of diabetic ketoacidemia. A 5-year prospective pediatric experience in 231 episodes. Arch Pediatr Adolesc Med. 1994; 148:1046-52.
22. Harris GD, Fiordalisi I, Harris WL, Mosovich LL, Finberg L. Minimizing the risk of brain herniation during treatment of diabetic ketoacidemia: a retrospective and prospective study. J Pediatr. 1990; 117:22-31.
23. Hanley JA, Lippman-Hand A. If nothing goes wrong, is everything all right? Interpreting zero numerators. JAMA. 1983; 249:1743-5.
24. Rother KI, Schwenk WF. Effect of rehydration fluid with 75 mmol/L of sodium on serum sodium concentration and serum osmolality in young patients with diabetic ketoacidosis. Mayo Clin Proc. 1994; 69:1149-53.
25. Rosenbloom AL, Riley WJ, Weber FT, Malone JI, Donnelly WH. Cerebral edema complicating diabetic ketoacidosis in childhood. J Pediatr. 1980; 96:357-61.
* a mole is defined as the molecular weight of the substance in grams, equal to Avogadro's number of molecules, 6.023x10^23. [return to text]
- Elliot J. Krane, M.D.
- E-mail: ejkrane@stanford.edu
- Departments of Anesthesia & Pediatrics
- Stanford University
- 300 Pasteur Blvd. Room H3584B
- Stanford, CA 94305-5115
- E-mail: ejkrane@stanford.edu